 2023, Vol. 35
2023, Vol. 35 


(2: 青海大学, 省部共建三江源生态与高原农牧业国家重点实验室, 西宁 810016)
(3: 青海大学, 黄河上游生态保护与高质量发展实验室, 西宁 810016)
(4: 青海大学, 水利部江河源区水生态治理与保护重点实验室, 西宁 810016)
(2: State Key Laboratory of Plateau Ecology and Agriculture, Qinghai University, Xining 810016, P. R. China)
(3: Provincial Laboratory of Ecological Protection and High Quality Development in the Upper Yellow River, Qinghai University, Xining 810016, P. R. China)
(4: Key Laboratory of Water Ecological Remediation and Protection at Headwater Regions of Big Rivers, Ministry of Water Resources, Qinghai University, Xining 810016, P. R. China)
沉积物(非流动相)和水体(流动相)是湖泊生态系统中两种不同但联系紧密的生境[1]。沉积物作为湖泊生态系统中重要的组成部分,一方面承载水体中元素(如:氮和磷)的沉降,另一方面,又为上层水体中生命体提供营养物质[2]。由于沉积物与水体的理化特征存在差异,湖泊生态系统中细菌群落特征也存在显著的差异[3-4]。Ren等研究黄河源区岩溶湖沉积物与水体中丰富和稀有细菌群落的差异时,发现沉积物中细菌alpha多样性显著低于水体,而beta多样性显著高于水体[5]。Song等研究我国新疆博斯腾湖与额碧湖细菌群落结构的结果表明,沉积物中细菌的多样性和丰度明显高于水体[6]。作为湖泊生态系统中重要的有机组分,细菌在湖泊生态系统物质转化和能量流动过程发挥着重要的作用[2]。因此,研究湖泊沉积物和水体细菌群落结构的差异对于湖泊水生态治理与保护具有重要的科学意义。
微生物的空间分布特点一定程度上代表了其在空间上的演替规律[7]。在环境选择和扩散限制的共同作用下,随着地理距离的增加,微生物群落结构的相似性呈线性减小的趋势,也称之为距离衰减关系(DDR),其拟合斜率的大小代表微生物群落在空间上周转率的大小[8]。不同生境中,环境选择(生境条件)和扩散限制(空间因子)对微生物空间分布的影响存在差异,进而导致不同生境中微生物的空间分布表现出明显的不同[9-11]。例如,Liu等研究长江水体浮游细菌和沉积物细菌的地理分布模式,发现水体和沉积物细菌空间分布格局差异较大[12]。然而,已有的研究大多关注低海拔湖泊生态系统中单一生境(水体或者沉积物)[13-14]或单一湖泊沉积物和水体中细菌群落特征[1],少有研究关注高海拔湖泊群沉积物和水体细菌群落空间分布及其差异。
基于相关关系的细菌共发生网络模式能够反映细菌群落之间复杂的相互作用、细菌群落构建以及细菌群落之间的生态位共享程度,在众多生境(土壤、湖泊、湿地和水库)相关研究中得到了应用[15-17]。Ji等发现青藏高原25个不同盐度的湖泊水体细菌群落共发生网络的复杂性随着盐度的增加而增加[18]。Yang等对比分析了柴达木盆地6个不同盐度的湖泊沉积物和水体细菌共发生网络的差异,发现水体细菌共发生网络的节点数、模块化、平均路径长度显著低于沉积物细菌共发生网络[19]。Ren等研究黄河源区喀斯特湖沉积物与水体细菌共发生网络的差异,发现沉积物细菌群落共发生网络较为聚集,而水体细菌群落共发生网络则较为复杂[20]。综合来看,先前关于青藏高原湖泊细菌群落共发生网络模式的研究均集中在小尺度上,缺乏对较大尺度湖泊群不同生境细菌共发生网络模式的解析。
揭示细菌群落构建过程是解析细菌多样性模式形成和维持的关键[21]。细菌群落的构建过程主要是选择、漂移、扩散、物种形成4个生态过程共同作用的结果[22],其中选择是一种确定性过程,使得细菌群落在相似的环境中趋于更相似的群落结构或非相似的环境中细菌群落结构的差异越大,漂移是导致细菌群落结构不确定性变化的随机性过程,扩散和物种形成是确定性过程和随机性过程共同作用的结果。虽然已有的研究结果表明,确定性过程和随机性过程共同驱动细菌群落的构建,但由于生境差异,流动性较强的生境(水体)细菌群落结构一般受随机性过程影响较大,而沉积物中大量颗粒物为细菌提供必须的营养物和能量则导致确定性过程对细菌群落构建过程贡献率较大[4, 24]。例如,Li等发现同向选择是主导三峡水库沉积物细菌群落构建的主导过程[23]。目前,针对青藏高原的湖泊微生物群落构建过程主要集中在青藏高原西部区域的水体生境中细菌和真核微生物的群落构建过程及其差异[25-26],而对青藏高原中东部湖泊沉积物和水体细菌群落构建过程的对比研究则相对较少。
本研究选择青藏高原中东部19个湖泊为研究对象,分别采集沉积物和水体样本后,基于高通量测序技术,解析沉积物和水体细菌多样性、群落结构及其驱动因素。结合采样点地理因子,研究采样湖泊沉积物和水体细菌的空间分布模式。此外,基于细菌OTUs之间相关性的大小构建细菌群落的共发生网络并计算其拓扑参数来反映网络结构的稳定性。最后,研究确定性过程和随机性过程对沉积物和水体细菌群落构建的相对贡献。本文关于青藏高原中东部湖泊沉积物与水体的细菌群落结构特征、空间分布模式、共发生网络模式和群落结构形成机制及其差异的研究成果,对深入理解高原湖泊细菌多样性和群落结构的格局形成具有重要的科学意义。
1 材料与方法 1.1 样本采集与处理2020年5—6月,选取青藏高原中东部19个湖泊共51个样点进行沉积物和水体样本的采集,采样湖泊和采样点地理位置信息见图 1和附表Ⅰ。使用水样采集器在每个采样点水下约0.5 m的位置采集水样5份放置在无菌窄口玻璃瓶中(约1 L),混匀后使用孔隙直径为0.22 μm的滤膜(47 mm,Millipore S-Pak Germany)进行抽真空过滤后收集滤膜。在同样的位置,使用抓斗式底泥采样器采集3个重复的表层沉积物样品,混匀后分装在50 mL无菌离心管中保存。将采集的102份样品均放置在便携式冰箱中-4℃暂存后送往实验室进行后续分析。

|
图 1 青藏高原采样湖泊分布 Fig.1 Locations of the sampling lakes on the Qinghai-Tibet Plateau |
表层沉积物样本的pH值采用GB 7859—1987标准测定,总氮采用GB 7848—1987标准测定,总磷采用GB 7852—1987标准测定。此外,使用AA3全自动连续流动分析仪对表层沉积物样本中氨氮和硝氮的浓度进行测定。水体理化参数的检测参考先前的研究[27]。沉积物样本和水体样本的检测结果分别见附表Ⅱ和附表Ⅲ。
1.3 DNA提取、PCR扩增和高通量测序将过滤后的滤膜和采集的沉积物样品按照CTAB法提取总DNA[28]。采用引物515F(5′-GTGCCAGCMGCCGCGGTTA-3′)/806R(5′-GGACTACHVGGGTWTCTAAT-3′)对细菌16S rRNA的高保真V4区进行PCR扩增[29]。PCR产物质检后送样进行双末端高通量测序(诺禾致源生物信息科技有限公司,北京)。
1.4 生物信息分析原始下机数据按如下标准化流程进行处理:(1)按照标签序列信息拆分各个样本测序数据后截取掉引物和标签序列获得每个样本短序列;(2)使用FLASH(v 1.2.7)对每个样本的短序列进行拼接,然后经过质控处理(位点截取、长度过滤、错配体和嵌合体去除)得到有效序列;(3)使用UPARSE软件(v7.0.1001)对所有样本的有效序列在97 % 的一致性水平上进行聚类得到OTUs;(4)基于SILVA138数据库对每个OTUs进行物种注释。原始测序数据已经上传至NCBI数据库,序列登记编号为PRJNA728144和PRJNA848449。
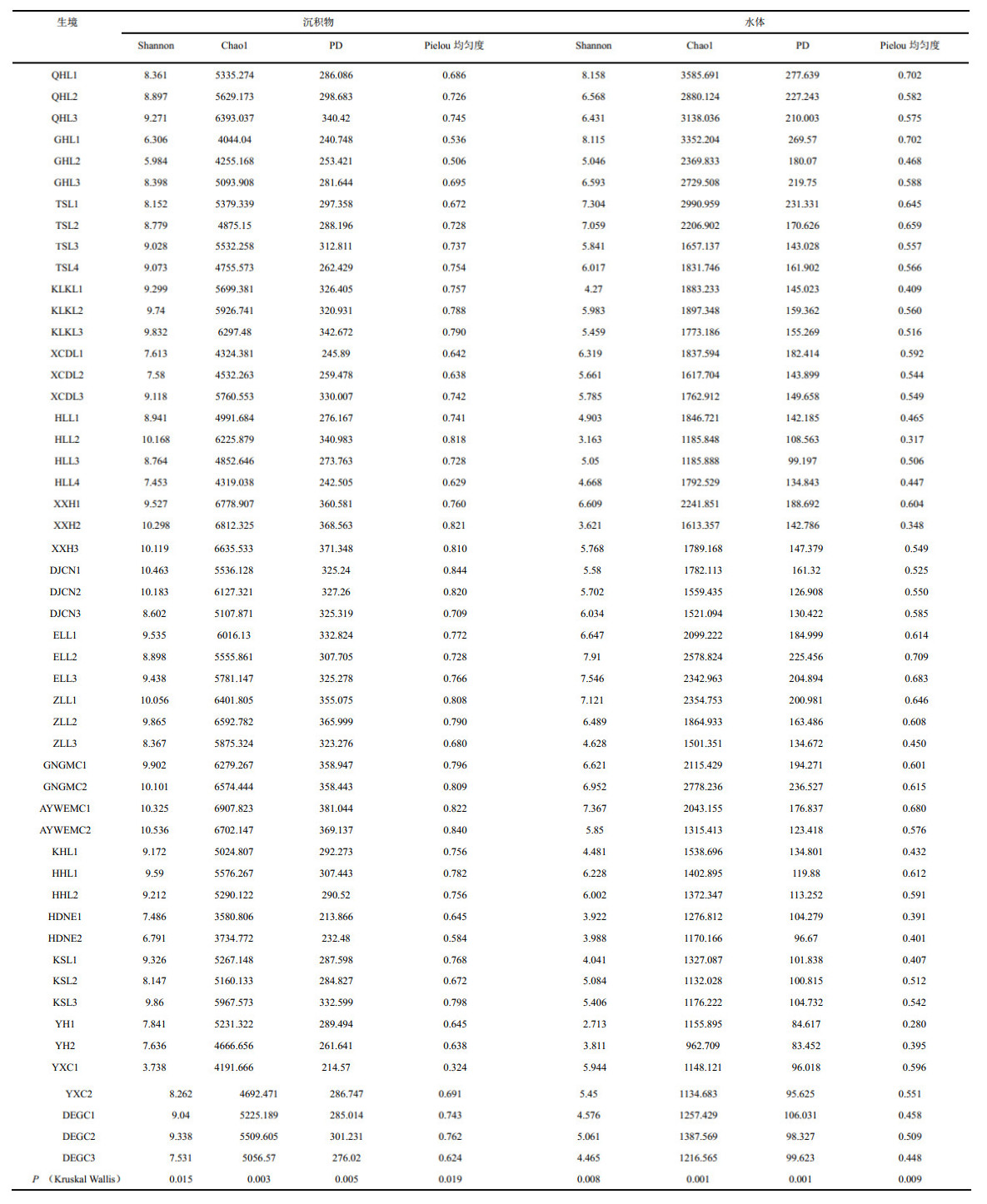
1.5 统计分析使用“vegan”和“picante”包计算沉积物和水体细菌群落的alpha多样性指数(Shannon指数、Chao1指数、Faith's系统发育指数和Pielou均匀度指数),并使用非参数Wilcoxon秩和检验分析沉积物和水体细菌群落alpha多样性指数的差异性[30]。使用Kruskal-Wallis检验分析不同湖泊沉积物/水体细菌群落alpha多样性指数的差异性。另外,基于细菌群落Bray-Curtis距离,使用主坐标分析(PCoA)研究水体和沉积物中细菌群落组成的差异,并使用相似性分析(ANOSIM)检测沉积物和水体细菌群落组成差异的显著性。基于OTU丰度表和方差膨胀因子(VIF)小于10的环境因子进行冗余分析(RDA),分别探究环境因子对沉积物和水体细菌群落结构变化的影响,同时使用“envfit”函数研究环境因子对沉积物和水体细菌群落结构影响的显著性[27]。通过线性拟合细菌群落的Bray-Curtis相似性指数与地理距离之间的关系,探讨沉积物和水体细菌群落的距离衰减模式[8]。
在OTU水平上构建沉积物和水体细菌群落的共发生网络模式。为了提高网络的准确性和可靠性,首先挑选出在超过一半数目样品(26个样品)中出现,并且其相对丰度大于0.01的OTUs,进一步选择Spearman相关系数绝对值大于0.8同时显著性P < 0.01的OTUs用于细菌群落共发生网络构建[1, 4]。使用Gephi(v 0.9.2)交互式软件对沉积物和水体共发生网络进行可视化。共发生网络的拓扑特征参数平均度(每个节点连接边数的平均值)、平均路径长度(每个节点连接其他节点的最短路径的平均值)、模块化(代表网络可以被划分为模块的程度,取值范围为0~1,模块化指数越高,表示网络可以划分的模块越多,反映网络复杂性越低,当模块化大于0.4时,认为网络具有模块化结构)、聚类系数(网络中节点倾向于聚集在一起的程度)使用“igraph”包进行计算[31-32]。节点的拓扑角色根据模块间(Pi)和模块内(Zi)连接值确定,当Pi≥0.62和Zi≥2.5时,节点为网络枢纽;当Pi < 0.62且Zi≥2.5时,节点为模块枢纽;当Pi≥0.62且Zi < 2.5时,节点为连接器;当Pi < 0.62且Zi < 2.5时,节点为外围节点[31]。一般而言,网络枢纽、模块枢纽和连接器被认为是构成整个细菌群落共发生网络的核心物种[20]。此外,反映水体和沉积物细菌网络结构稳定性的鲁棒性和脆弱性两个参数根据参考文献中的脚本进行计算[33-34]。
使用“spaa”包的niche.width函数计算沉积物和水体细菌的Levins'生态位宽度[35]。基于OTU丰度表,使用中性模型评估高原湖泊沉积物与水体细菌菌群的迁移率值[27]。为了量化确定性过程和随机性过程对沉积物和水体中细菌群落构建的贡献,使用“nst”包对基于Bray-Curtis距离的归一化随机率(NST)进行计算[36]。当NST>0.5时,随机性过程主导细菌群落的构建过程,相反地,当NST < 0.5时,认为确定性过程主导细菌群落的构建[1, 36]。为了进一步量化沉积物和水体细菌群落构建过程,使用“iCAMP”包的icamp.big函数计算同向选择、异向选择、同向扩散、扩散限制和漂变分别对沉积物和水体细菌群落构建过程的相对贡献[37]。以上所有数据分析均在R软件(v 4.0.5)中进行。
2 结果分析 2.1 沉积物与水体理化因子差异采样湖泊所有沉积物样本和水体样本均为碱性(沉积物pH平均值为9.07,水体pH为9.40)。高原湖泊沉积物中氨氮、硝氮和总磷浓度明显高于水体,其中沉积物中氨氮、硝态氮和总磷含量的平均值分别为11.52、5.81和1.08 g/kg(附表Ⅱ),水体中氨氮、硝态氮和总磷的平均浓度为0.12、0.19和0.04 mg/L(附表Ⅲ)。沉积物和水体总氮分别为0.19~6.15 g/kg和0.52~2.50 mg/L(附表Ⅱ,Ⅲ)。
2.2 细菌群落特征和空间分布差异所有采样湖泊沉积物样本测序分析后共得到OTUs的数目为99541,分别隶属于92门1169属。所有水体样本测序共得到OTUs的数目为34269,分别隶属于83门1036属。沉积物和水体细菌alpha多样性指数计算结果如附表Ⅳ所示。不同湖泊的沉积物/水体的细菌群落alpha多样性指数差异显著(Kruskal-Wallis P < 0.05),其中阿涌吾儿玛措湖(AYWEMC)沉积物细菌的Shannon多样性指数(10.43)和Chao1指数(6804.99)最大,而鄂陵湖(ELL)水体细菌的Shannon多样性指数(7.37)和Chao1指数(2340.34)最大。非参数Wilcoxon秩和检验结果表明,沉积物细菌群落Chao1指数、Shannon多样性指数、Faith's系统发育指数和Pielou均匀度指数均显著高于水体细菌群落(P < 0.001),这表明沉积物细菌群落由更为多样和复杂的物种组成(图 2)。

|
图 2 沉积物与水体细菌群落alpha多样性指数对比分析 Fig.2 Comparition of alpha diverisity indices of bacterial communities between sediment and water |
采样湖泊沉积物和水体中细菌的主要门类基本相似,平均相对丰度前4的门类均为变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和放线菌门(Actinobacteria),但其平均相对丰度在沉积物和水体中明显不同。在沉积物中,Proteobacteria的平均相对丰度为45.33 %,Bacteroidetes为14.59 %,Firmicutes为8.07 %,Actinobacteria为5.92 %。在水体中,Proteobacteria的平均相对丰度为41.77 %,Bacteroidetes为28.21 %,Firmicutes为4.65 %,Actinobacteria为11.19 % (图 3)。可以看出Proteobacteria、Bacteroidetes和Actinobacteria表现出一定的沉积物偏好性。在纲分类水平上,沉积物中细菌的优势菌为Gammaproteobacteria,平均相对丰度占比为32.84 %,水体中细菌的优势菌为Bacteroidia(28.85 %)(图 3)。进一步使用PCoA和ANOSIM对沉积物和水体细菌群落结构的差异性进行解析,结果如图 4所示。由图 4可知,沉积物和水体细菌群落呈现出显著差异,且不同湖泊沉积物中细菌群落分布较为聚集,水体细菌群落结构分布较为分散,这表明青藏高原湖泊沉积物细菌群落空间同质性高,而水体细菌群落空间异质性高。PCoA1和PCoA2对沉积物和水体细菌差异的贡献率为24.65 %,沉积物和水体的细菌群落结构存在极显著的差异(ANOSIM R=0.645,P=0.001)。RDA结果表明海拔是青藏高原湖泊沉积物细菌群落结构的最显著驱动因子。水体环境因子对其细菌群落结构的影响大小为:纬度>pH>溶解氧,其中,纬度是青藏高原湖泊水体细菌群落结构最显著的驱动因子(图 5)。RDA1与RDA2之和分别解释沉积物和水体细菌群落结构变化的20.12 % 和26.81 %。

|
图 3 不同湖泊沉积物和水体细菌群落中相对丰度前10的门(a、b)和纲(c、d) (缩写对应的湖泊名称见附表Ⅰ) Fig.3 Top 10 phyla (a, b) and classes (c, d) of bacterial communities in sediment and water of different lakes |

|
图 4 基于Bray-Curtis距离的沉积物与水体细菌群落主坐标分析 Fig.4 Principal coordinate analysis of bacterial communities in sediment and water based on the Bray-Curtis distance |

|
图 5 沉积物和水体细菌群落结构的冗余分析 (带*的环境参数代表envfit函数检验P < 0.05,**代表P < 0.01, ***代表P < 0.001;海拔-Altitude;纬度-Latitude;经度-Longitude;沉积物总磷-SediTP;沉积物铵态氮-SediNH4+-N;沉积物硝态氮-SediNO3--N;沉积物pH-Sedi pH;水体总氮-Wat TN;水体pH-Wat pH;水体硝态氮-WatNO3--N;溶解氧-DO;盐度-SAL) Fig.5 Redundancy analysis of bacterial community structure in sediment and water (Environmental with * represents the P-value < 0.05 using function of envfit test, ** represents the P-value < 0.01, *** represents the P-value < 0.001) |
细菌群落的Bray-Curtis相似性指数与地理距离的线性拟合结果揭示了采样湖泊沉积物和水体细菌群落均具有明显的距离衰减模式(图 6),水体细菌群落距离衰减模式与沉积物相比具有更大的斜率(绝对值分别为0.18和0.07),这表明水体细菌群落在空间上的周转率大于沉积物细菌群落。

|
图 6 沉积物和水体细菌群落Bray-Curtis相似性指数与地理距离之间的关系(R2代表矫正后的拟合优度) Fig.6 Relationship between Bray-Curtis similarity of bacterial communities and geographical distance in sediment and water (R2 represents fitness coefficient after correction) |
在OTU水平上构建沉积物和水体细菌的共发生网络如图 7所示,其对应的网络拓扑参数见表 1。由图 7和表 1可见,沉积物和水体细菌群落共发生网络存在明显的差异。表 1中沉积物细菌共发生网络由450个节点和2191条边组成,水体细菌群落共发生网络由223个节点和684条边组成,表明青藏高原湖泊沉积物细菌OTUs之间具有更复杂的互联关系和更复杂的网络结构。沉积物和水体细菌群落共发生网络模式的模块化分别为0.651和0.675(P>0.4),这表明沉积物和水体细菌均具有较高的模块化结构,但沉积物细菌共发生网络的模块化略小于水体细菌,说明水体细菌共发生网络的模块之内的连接较为复杂,而沉积物细菌共发生网络的模块间连接较为复杂。此外,沉积物细菌的共发生网络模式的平均聚类系数和节点的平均度均明显高于水体细菌,而模块化低于水体,这表明在环境干扰下沉积物细菌共发生网络的稳定性更高。另外,沉积物和水体细菌网络的鲁棒性指数值分别为0.382和0.349,脆弱性指数分别为0.039和0.188(表 1),也同样表明,沉积物细菌群落共发生网络的稳定性要高于水体细菌。

|
图 7 沉积物和水体细菌群落共发生网络 (按照不同的模块进行着色,仅展示网络中相对丰度前5的模块,其他模块用灰色表示;节点大小代表平均连接度,颜色代表不同的模块;边的颜色代表不同的模块) Fig.7 Co-occurrence networks of bacterial communities in sediment and water (only the relative abundance of top 5 modules with different colors are displayed based on their modularity, and other modules are represented with grey; size of nodes represents average degree, color of nodes represents different modularity; color of edges indicates different modularity) |
| 表 1 沉积物和水体细菌共发生网络拓扑参数 Tab. 1 Topological properties of bacterial co-occurrence networks in sediment and water |
根据沉积物和水体共发生网络模式中模块间(Pi)和模块内(Zi)连接值大小,确定各自节点的拓扑角色,即其属于网络枢纽、模块枢纽、连接器,还是外围节点,由此确定相应的核心物种(图 8)。从图 8可以看出,沉积物细菌群落共发生网络模式中核心物种数目明显多于水体,其中沉积物中的核心物种主要为Acidobacteria、Burkholderiaceae、Nitrosomonadaceae、Rhodobacteraceae、Loktanella。水体中的核心物种为Sporichthyaceae和Pseudarcicella。

|
图 8 基于网络拓扑角色的沉积物与水体细菌的核心物种鉴定 Fig.8 Identified keystone species of bacterial in sediment and water based on the topological roles in networks |
采样湖泊沉积物和水体细菌群落的Levins'生态位宽度(Bcom)计算结果如图 9所示。沉积物细菌群落的平均生态位宽度值为90.38,显著高于水体细菌(17.54,P < 0.001),这表明与生态位有关的确定性过程对沉积物细菌群落的影响更大。为了进一步量化随机性过程和确定性过程对采样湖泊沉积物和水体细菌群落构建的相对贡献,基于Bray-Curtis距离计算了沉积物和水体细菌群落的NST分别为0.24和0.72(图 9)。沉积物细菌群落的NST显著低于水体细菌群落(P < 0.001),这表明沉积物和水体中细菌群落的构建过程显著不同,其中确定性过程对青藏高原湖泊沉积物细菌群落构建起到了主导,而随机性过程则对青藏高原湖泊水体细菌群落构建过程中更为重要。使用iCAMP进一步量化不同生态过程对沉积物和水体细菌群落构建过程的相对贡献率,结果如表 2所示。随机性过程(同向扩散、扩散限制和漂变之和)对青藏高原湖泊沉积物和水体细菌群落构建的相对贡献率分别为49.64 % 和56.43 %,确定性过程(同向选择与异向选择之和)对采样湖泊沉积物和水体细菌群落的相对贡献率分别为50.36 % 和43.48 %,这进一步证实了确定性过程对青藏高原湖泊沉积物细菌群落贡献率更高,而随机性过程在水体细菌群落构建过程中占主导的结论。

|
图 9 沉积物和水体细菌群落的平均生态位宽度和归一化随机率值 Fig.9 The mean habitat niche breadth from all taxa for samples (Bcom) and value of normalized stochasticity ratio of bacterial communities in sediment and water |
| 表 2 不同生态过程对沉积物和水体细菌群落构建过程的相对重要性 Tab. 2 Relative importance of different ecological processes of bacterial communities in sediment and water |
本研究中采样的19个湖泊沉积物和水体在细菌多样性、群落结构、空间分布、共发生网络模式、群落构建过程方面存在极显著的差异。
3.1 沉积物和水体细菌群落特征差异显著本研究中青藏高原湖泊沉积物细菌群落多样性显著高于水体细菌群落,这与先前报道的青藏高原柴达木盆地不同盐度的湖泊群沉积物原核微生物alpha多样性显著高于水体的结果一致[38]。研究发现超过60 % 以上的细菌都趋于与悬浮/沉降的颗粒物粘连,而沉积物中固体颗粒物能够为细菌提供必要的附着体和充足的营养物,这是导致沉积物细菌Chao1和Shannon多样性指数高于水体的原因[39]。另外,沉积物细菌群落更宽的生态位(图 9),也可能是沉积物细菌的Chao1和Shannon多样性指数高于水体细菌的原因。根据生态位的互补性,更宽的微生物群落生态位能够减少其物种之间对资源的竞争,有利于不同物种的共存,而使沉积物细菌多样性高于水体[35]。然而,采样湖泊沉积物和水体细菌群落多样性的差异与Ren等研究黄河源区岩溶湖沉积物细菌alpha多样性低于水体细菌的结论不一致,可能由于在其研究的熔岩湖中热熔岩湖泊沉积物中高浓度的石灰岩对细菌细胞造成了物理损害或者限制其生长繁殖[5]造成了这一差异。
尽管采样湖泊沉积物和水体细菌的群落结构显著不同,但湖泊沉积物和水体细菌的优势门均为Proteobacteria,这与先前研究青藏高原湖泊沉积物或者水体生境中群落结构的结果相一致[40-42]。虽然不同湖泊的沉积物细菌均以Proteobacteria为主(平均相对丰度为41.77 %),但一些采样湖泊的水体优势菌表现出差异,其中星星海和扎陵湖以Cyanobacteria为优势菌门(39.76 % 和39.73 %),而海丁诺尔、盐湖和多尔改措物种相对丰度最高的是Bacteroidetes,相对丰度分别为52.66 %、56.09 % 和50.54 % (图 3),采样湖泊沉积物与水体细菌群落结构在空间异质性方面存在差异。究其原因,一方面由于湖泊水体比沉积物更易受到周围动物活动的影响且水体受到外界干扰的程度不一,如受到动物活动影响较大的克鲁克湖水体细菌组成中Firmicutes相对丰度占比最高[42];另一方面,由于Cyanobacteria门中有些类群的细胞能够形成调节自身浮力的气泡,使得其在水体生境中与其他门类在竞争营养盐和光热的方面具有明显的优势,使得Cyanobacteria在部分湖泊水体中富集[43]。
3.2 沉积物和水体细菌空间分布模式和共发生网络模式差异显著一般情况下,水体的流动性会减弱其对细菌群落的扩散限制使细菌群落在空间上的周转率降低,而相对静止的沉积物生境能增加其对细菌菌群的扩散限制使细菌群落的空间周转率增大,最终表现为水体中细菌群落空间周转率往往小于沉积物细菌菌群的空间周转率[44]。但是,本研究中采样湖泊沉积物细菌空间周转率(距离衰减拟合斜率绝对值)明显小于水体细菌,可能的原因如下:1)本研究中不同湖泊沉积物和水体细菌群落结构差异性分析结果(图 4)表明,水体细菌群落结构在相同空间尺度上的分布具有更高的异质性,说明采样湖泊水体细菌更易受到自然和周围动植物因素的影响,外界营养源对水体细菌群落结构的影响作用强于沉积物,进而表现为水体细菌菌群空间周转率大于沉积物[1]。2)可能采样湖泊沉积物细菌群落更均匀的分布(图 2),使其在空间上的分类学差异减小,表现为沉积物细菌群落空间周转率减小。3)采样湖泊水体中细菌群落的构建相比沉积物更多地受随机性过程主导(图 9),进一步使用Sloan等的中性模型评估了沉积物和水体细菌菌群的迁移率值,其细菌迁移率值分别为0.27和0.08[45]。因此,推测水体细菌菌群本身扩散能力较弱,受到的扩散限制作用更强,导致水体细菌群落在空间上周转率增大,这一推断也可以从表 2相关数据中得到证实。
在共发生网络中,模块更可能具有独立功能的生态位[18, 39]。采样湖泊沉积物和水体两种不同的生境共发生网络模式也存在差异,这与Yang等针对柴达木盆地湖泊的研究结果基本一致[19]。本研究中采样湖泊沉积物细菌群落共发生网络模块数多于水体细菌,表明沉积物细菌群落比水体细菌群落具有更多样的生态位和复杂的功能[46],此差异归因于沉积物中更多样和丰富的细菌群落,使细菌菌群之间产生了更复杂的相互作用(共生、竞争、捕食)而形成了更复杂的网络结构[20]。
另外,关键物种因其强烈而独特的种间相互作用对整个细菌群落的稳定性做出了巨大的贡献,一旦关键物种消失可能导致整个模块甚至整个网络的崩溃[32]。同样地,本研究中采样湖泊沉积物细菌群落具有更多核心物种(图 8)和更稳定的网络结构(更高的网络鲁棒性和更低的网络脆弱性)(表 1),这可能是由于沉积物与水体之间的生境差异较大(附表Ⅱ,附表Ⅲ),从而引起细菌群落的网络结构和拓扑特征的差异[44]。
3.3 沉积物和水体细菌群落构建差异显著确定性过程和随机性过程被认为是驱动微生物多样性和分布格局形成的最重要过程。近年来,基于生态位理论与中性理论的中性模型和零模型解析了众多生境(河流、湖泊、土壤等)微生物群落的构建机理[44, 47-48]。本研究中使用归一化随机率和iCAMP框架研究青藏高原湖泊沉积物和水体细菌群落构建过程及其差异,结果均表明确定性过程为沉积物细菌群落的主导构建过程,而主导水体细菌群落构建的过程为随机性过程。生态位理论认为环境和生物过滤等确定性过程会影响细菌群落构建,而本研究中计算得到的沉积物细菌群落的平均生态位宽度显著大于水体,说明沉积物细菌群落相比于水体更易受到环境挑选的影响,从而表现其构建过程受确定性过程主导。先前的一项研究指出了与水体相比,更加异质和含有大量微生境的沉积物可以为细菌提供不同的生境条件,增加了确定性过程对其细菌群落的贡献[4]。这是由于水体中细菌菌群扩散能力较低,使得水体细菌更易受到随机过程(扩散限制)的影响(表 2)。另外,由于采样湖泊沉积物中细菌群落丰度和多样性高于水体细菌群落而导致的沉积物细菌菌群之间的相互作用增强,也使确定性过程在沉积物细菌群落构建过程中的贡献增加[1, 4]。
4 结论本文选取了青藏高原中东部共19个湖泊为研究对象,系统解析了采样湖泊沉积物和水体细菌群落结构、空间分布模式、共发生网络模式、群落构建过程及其差异,并讨论了差异形成的原因。论文取得的主要研究结论如下:(1)由于采样湖泊沉积物能够为细菌提供更多的生态位,使沉积物细菌群落的Chao1和Shannon多样性指数显著高于水体细菌群落。Proteobacteria均为沉积物和水体细菌的优势菌门(沉积物:45.33 %;水体:41.77 %),但沉积物和水体细菌群落结构存在极显著差异(ANOSIM R=0.645,P=0.001),同时水体细菌群落结构表现出较强的空间异质性。海拔和纬度分别是青藏高原湖泊沉积物和水体细菌群落结构变化最显著的驱动因子;(2)采样湖泊沉积物和水体细菌群落均具有显著的距离衰减模式,水体细菌群落在空间上的周转率大于沉积物细菌群落;(3)采样湖泊沉积物细菌群落共发生网络复杂度和稳定性高于水体细菌,表明在环境干扰下,采样湖泊沉积物细菌群落网络更稳定;(4)采样湖泊沉积物细菌群落构建过程主要为确定性过程,而随机性过程在水体细菌群落构建中占据主导地位,表明水体细菌群落相比于沉积物细菌群落更易受到扩散限制的影响,而沉积物细菌群落之间更复杂的相互作用增加了确定性过程对其细菌群落构建过程的贡献。
5 附录附表Ⅰ~Ⅳ见电子版(DOI: 10.18307/2023.0316)。
| 附表Ⅰ 采样点的空间分布 Appendix Ⅰ Geographical distributions of sampling sites |

| 附表Ⅱ 不同样点沉积物物理化参数 Appendix Ⅱ Physicchemical factors of sediment in different sampling sites |

| 附表Ⅲ 不同样点水体物理化参数 Appendix Ⅲ Physicchemical factors of water in different sampling sites |
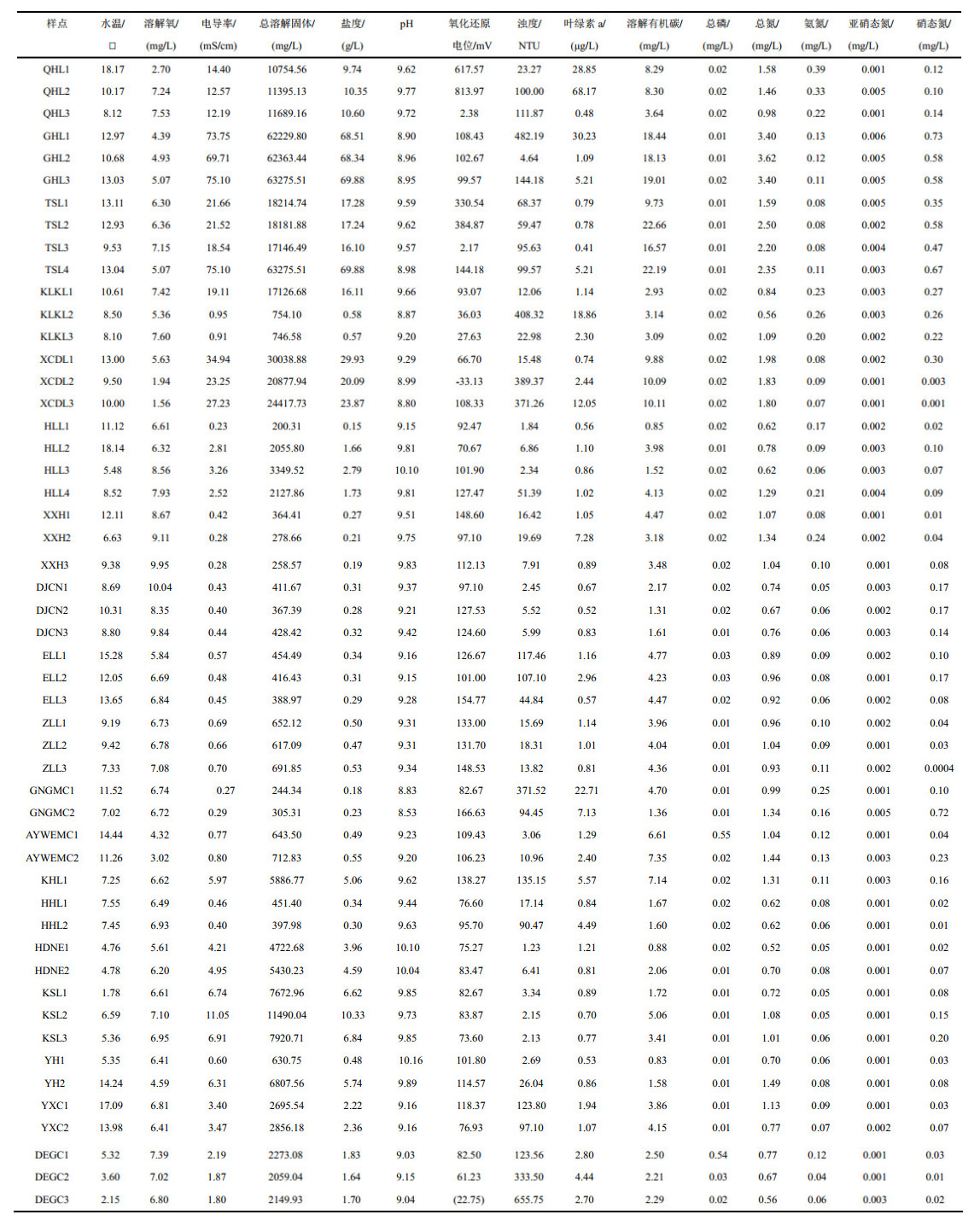
| 附表Ⅳ 沉积物和水体不同样点细菌群落alpha多样性 Appendix Ⅳ Alpha diversity indices of bacterial community in different sampling points in sediment and water |
| [1] |
Zeng J, Jiao CC, Zhao DY et al. Patterns and assembly processes of planktonic and sedimentary bacterial community differ along a trophic gradient in freshwater lakes. Ecological Indicators, 2019, 106: 105491. DOI:10.1016/j.ecolind.2019.105491 |
| [2] |
孙芳. 蓝藻水华水体底泥微生物群落结构及铜锈环棱螺生物干扰的影响[学位论文]. 宁波: 宁波大学, 2012.
|
| [3] |
孙旭. 富营养化湖泊底栖动物对沉积物氮转化菌群落结构和功能的影响[学位论文]. 南京: 南京大学, 2015.
|
| [4] |
Jiao CC, Zhao DY, Zeng J et al. Disentangling the seasonal co-occurrence patterns and ecological stochasticity of planktonic and benthic bacterial communities within multiple lakes. Science of the Total Environment, 2020, 740: 140010. DOI:10.1016/j.scitotenv.2020.140010 |
| [5] |
Ren Z, Zhang C, Li X et al. Abundant and rare bacterial taxa structuring differently in sediment and water in thermokarst lakes in the Yellow River Source area, Qinghai-Tibet Plateau. BioRxiv, 2021. DOI:10.1101/2021.05.26.445882 |
| [6] |
Song WJ, Qi R, Zhao L et al. Bacterial community rather than metals shaping metal resistance genes in water, sediment and biofilm in lakes from arid northwestern China. Environmental Pollution, 2019, 254: 113041. DOI:10.1016/j.envpol.2019.113041 |
| [7] |
Li L, Ning DL, Jeon Y et al. Ecological insights into assembly processes and network structures of bacterial biofilms in full-scale biologically active carbon filters under ozone implementation. Science of the Total Environment, 2021, 751: 141409. DOI:10.1016/j.scitotenv.2020.141409 |
| [8] |
Milici M, Tomasch J, Wos-Oxley ML et al. Bacterioplankton biogeography of the Atlantic Ocean: A case study of the distance-decay relationship. Frontiers in Microbiology, 2016, 7: 590. DOI:10.3389/fmicb.2016.00590 |
| [9] |
Zeng QC, An SS, Liu Y et al. Biogeography and the driving factors affecting forest soil bacteria in an arid area. Science of the Total Environment, 2019, 680: 124-131. DOI:10.1016/j.scitotenv.2019.04.184 |
| [10] |
Liu JW, Zhu SQ, Liu XY et al. Spatiotemporal dynamics of the archaeal community in coastal sediments: Assembly process and co-occurrence relationship. The ISME Journal, 2020, 14(6): 1463-1478. DOI:10.1038/s41396-020-0621-7 |
| [11] |
Zhang HJ, Huang XL, Huang L et al. Microeukaryotic biogeography in the typical subtropical coastal waters with multiple environmental gradients. Science of the Total Environment, 2018, 635: 618-628. DOI:10.1016/j.scitotenv.2018.04.142 |
| [12] |
Liu T, Zhang AN, Wang JW et al. Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome, 2018, 6(1): 16. DOI:10.1186/s40168-017-0388-x |
| [13] |
Yi YJ, Lin CQ, Wang WJ et al. Habitat and seasonal variations in bacterial community structure and diversity in sediments of a Shallow Lake. Ecological Indicators, 2021, 120: 106959. DOI:10.1016/j.ecolind.2020.106959 |
| [14] |
Chen L, Hu BX, Dai H et al. Characterizing microbial diversity and community composition of groundwater in a salt-freshwater transition zone. Science of the Total Environment, 2019, 678: 574-584. DOI:10.1016/j.scitotenv.2019.05.017 |
| [15] |
Liu Z, Ma XM, He N et al. Shifts in microbial communities and networks are correlated with the soil ionome in a kiwifruit orchard under different fertilization regimes. Applied Soil Ecology, 2020, 149: 103517. DOI:10.1016/j.apsoil.2020.103517 |
| [16] |
Qin MY, Xu HM, Zhao DY et al. Aquaculture drives distinct patterns of planktonic and sedimentary bacterial communities: Insights into co-occurrence pattern and assembly processes. Environmental Microbiology, 2022, 24(9): 4079-4093. DOI:10.1111/1462-2920.15922 |
| [17] |
Wang BH, Zheng XF, Zhang HJ et al. Keystone taxa of water microbiome respond to environmental quality and predict water contamination. Environmental Research, 2020, 187: 109666. DOI:10.1016/j.envres.2020.109666 |
| [18] |
Ji MK, Kong WD, Yue LY et al. Salinity reduces bacterial diversity, but increases network complexity in Tibetan Plateau lakes. FEMS Microbiology Ecology, 2019, 95(12): fiz190. DOI:10.1093/femsec/fiz190 |
| [19] |
Yang J, Jiang HC, Sun XX et al. Distinct co-occurrence patterns of prokaryotic community between the waters and sediments in lakes with different salinity. FEMS Microbiology Ecology, 2020, 97(1): fiaa234. DOI:10.1093/femsec/fiaa234 |
| [20] |
Ren Z, Zhang C, Li X et al. Bacterial communities present distinct co-occurrence networks in sediment and water of the thermokarst lakes in the Yellow River source area. Frontiers in Microbiology, 2021, 12: 716732. DOI:10.3389/fmicb.2021.716732 |
| [21] |
Chen WD, Ren KX, Isabwe A et al. Correction to: Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome, 2019, 7: 148. DOI:10.1186/s40168-019-0749-8 |
| [22] |
Vellend M. Conceptual synthesis in community ecology. The Quarterly Review of Biology, 2010, 85(2): 183-206. DOI:10.1086/652373 |
| [23] |
Li Y, Gao Y, Zhang WL et al. Homogeneous selection dominates the microbial community assembly in the sediment of the Three Gorges Reservoir. Science of the Total Environment, 2019, 690: 50-60. DOI:10.1016/j.scitotenv.2019.07.014 |
| [24] |
He Q, Wang S, Hou WG et al. Temperature and microbial interactions drive the deterministic assembly processes in sediments of hot springs. Science of the Total Environment, 2021, 772: 145465. DOI:10.1016/j.scitotenv.2021.145465 |
| [25] |
Gu ZQ, Liu KS, Pedersen MW et al. Community assembly processes underlying the temporal dynamics of glacial stream and lake bacterial communities. Science of the Total Environment, 2021, 761: 143178. DOI:10.1016/j.scitotenv.2020.143178 |
| [26] |
Liu KS, Liu YQ, Hu AY et al. Different community assembly mechanisms underlie similar biogeography of bacteria and microeukaryotes in Tibetan lakes. FEMS Microbiology Ecology, 2020, 96(6): fiaa071. DOI:10.1093/femsec/fiaa071 |
| [27] |
Wang DD, Huang YF, Yang HJ. Seasonal differences of lakes bacterial community structure and driving mechanism at the northeastern of the Qinghai-Tibet Plateau. J Lake Sci, 2023, 35(1): 267-282. [王丹丹, 黄跃飞, 杨海娇. 青藏高原东北部湖泊细菌群落结构特征季节性差异及驱动机制. 湖泊科学, 2023, 35(1): 267-282. DOI:10.18307/2023.0121] |
| [28] |
Shen L, Yao TD, Xu BQ et al. Variation of culturable bacteria along depth in the East Rongbuk ice core, Mt. Everest. Geoscience Frontiers, 2012, 3(3): 327-334. DOI:10.1016/j.gsf.2011.12.013 |
| [29] |
Caporaso JG, Lauber CL, Walters WA et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Journal of Microbiological Methods, 2011, 108(Suppl 1): 4516-4522. DOI:10.1073/pnas.1000080107 |
| [30] |
Caporaso JG, Kuczynski J, Stombaugh J et al. QⅡME allows analysis of high-throughput community sequencing data. Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303 |
| [31] |
Deng Y, Jiang YH, Yang YF et al. Molecular ecological network analyses. BMC Bioinformatics, 2012, 13: 113. DOI:10.1186/1471-2105-13-113 |
| [32] |
Tu QC, Yan QY, Deng Y et al. Biogeographic patterns of microbial co-occurrence ecological networks in six American forests. Soil Biology and Biochemistry, 2020, 148: 107897. DOI:10.1016/j.soilbio.2020.107897 |
| [33] |
Liu SW, Yu H, Yu YH et al. Ecological stability of microbial communities in Lake Donghu regulated by keystone taxa. Ecological Indicators, 2022, 136: 108695. DOI:10.1016/j.ecolind.2022.108695 |
| [34] |
Yuan MM, Guo X, Wu LW et al. Climate warming enhances microbial network complexity and stability. Nature Climate Change, 2021, 11(4): 343-348. DOI:10.1038/s41558-021-00989-9 |
| [35] |
Chen QL, Hu HW, Yan ZZ et al. Deterministic selection dominates microbial community assembly in termite mounds. Soil Biology and Biochemistry, 2021, 152: 108073. DOI:10.1016/j.soilbio.2020.108073 |
| [36] |
Liu SF, Chen Q, Li JR et al. Different spatiotemporal dynamics, ecological drivers and assembly processes of bacterial, archaeal and fungal communities in brackish-saline groundwater. Water Research, 2022, 214: 118193. DOI:10.1016/j.watres.2022.118193 |
| [37] |
Ning DL, Yuan MT, Wu LW et al. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nature Communications, 2020, 11: 4717. DOI:10.1038/s41467-020-18560-z |
| [38] |
Yang J, Ma LA, Jiang HC et al. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Scientific Reports, 2016, 6: 25078. DOI:10.1038/srep25078 |
| [39] |
Lv BY, Shi JH, Li T et al. Deciphering the characterization, ecological function and assembly processes of bacterial communities in ship ballast water and sediments. Science of the Total Environment, 2022, 816: 152721. DOI:10.1016/j.scitotenv.2021.152721 |
| [40] |
Liu KS, Hou JZ, Liu YQ et al. Biogeography of the free-living and particle-attached bacteria in Tibetan lakes. FEMS Microbiology Ecology, 2019, 95(7): 1-10. DOI:10.1093/femsec/fiz088 |
| [41] |
Liu KS, Yao TD, Pearce DA et al. Bacteria in the lakes of the Tibetan Plateau and polar regions. Science of the Total Environment, 2021, 754: 142248. DOI:10.1016/j.scitotenv.2020.142248 |
| [42] |
Zhang L, Fang WK, Li XC et al. Linking bacterial community shifts with changes in the dissolved organic matter pool in a eutrophic lake. Science of the Total Environment, 2020, 719: 137387. DOI:10.1016/j.scitotenv.2020.137387 |
| [43] |
陈楠. 太湖沉积物微生物群落组成与物质循环及藻华爆发的相关性[学位论文]. 北京: 中国农业大学, 2015.
|
| [44] |
He JT, Zhang N, Muhammad A et al. From surviving to thriving, the assembly processes of microbial communities in stone biodeterioration: A case study of the West Lake UNESCO World Heritage area in China. Science of the Total Environment, 2022, 805: 150395. DOI:10.1016/j.scitotenv.2021.150395 |
| [45] |
Sloan WT, Lunn M, Woodcock S et al. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environmental Microbiology, 2006, 8(4): 732-740. DOI:10.1111/j.1462-2920.2005.00956.x |
| [46] |
Ezeokoli OT, Nuaila VNA, Obieze CC et al. Assessing the impact of rice cultivation and off-season period on dynamics of soil enzyme activities and bacterial communities in two agro-ecological regions of Mozambique. Agronomy, 2021, 11(4): 694. DOI:10.3390/agronomy11040694 |
| [47] |
Liu B, Yao J, Chen ZH et al. Biogeography, assembly processes and species coexistence patterns of microbial communities in metalloids-laden soils around mining and smelting sites. Journal of Hazardous Materials, 2022, 425: 127945. DOI:10.1016/j.jhazmat.2021.127945 |
| [48] |
Tripathi BM, Stegen JC, Kim M et al. Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. The ISME Journal, 2018, 12(4): 1072-1083. DOI:10.1038/s41396-018-0082-4 |